
易算生物今天给你带来一篇宏蛋白组学的新文章。新的一年里,祝大家挖到大矿,深度挖宝。
摘要
历经了多年发展,宏蛋白质组学已经显示出这两方面突破:能够揭示复杂微生物群落(如肠道微生物群)的分类组成;还能够揭示有关动态宿主-微生物群相互作用的深层功能信息。
但是,宏蛋白质组中的准确蛋白质鉴定和定量才是真正的分水岭:这是由肠道代谢蛋白质组的极端复杂性和异质性所决定。怎么解决?这篇文章以及文末的引用文献一定可以帮到你。
农历新年我们也没有停下前进的脚步,复旦大学、上海交通大学医学院、长海医院与易算生物携手发表在npj|biofilms and microbiomes上的最新文章:
Data-independent acquisition boosts quantitative metaproeomics for deep characterization of gut microbiota。为DIA宏蛋白质组从技术上成为解决相关生物问题的核心手段进一步打下基础。
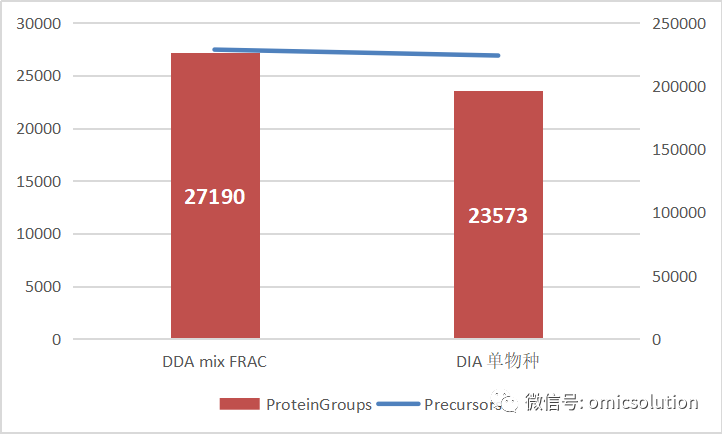
在我们最新获取的数据中,1小时DIAPASEF可以从12个物种细菌的混合物中定量得到超过20000个Protein Group,从而证明DIA在超复杂蛋白质体系中进行蛋白质定量的极高效率。
敲黑板:宏蛋白质组研究中目前存在这三个技术难点:
蛋白种类过于复杂,检测深度远远小于实际蛋白组成;
上一个问题带来的定量准确性难以保证;
物种组成复杂,可定量的物种独有肽段少,物种区分准确性低;
假如你的研究受限于上述我提到的,那么接下来的内容会很适合你。本文则致力于证明前两个问题可以采用DIA技术解决,并进一步间接改善第3个问题。
文章简介简介

在过去几年中,虽然大多数蛋白质组学和宏蛋白质组学研究采用基于数据依赖性采集 (DDA) 的传统质谱 (MS) 方法,但数据非依赖性采集 (DIA) 技术在传统蛋白质组学中表现出色,蛋白质组覆盖率、重现性和定量准确性更高【1】。
2020年,Aakko等人率先在宏蛋白质组学中使用DIA,并报告了一项概念验证研究,以证明其技术可行性【2】。同年,复旦乔亮团队联合易算还报告了最早的基于DIA的宏蛋白质组学研究之一,其中在真实的临床队列中从肠道微生物群中鉴定和量化了30000多种蛋白质【3】。
作为一种新兴技术,基于DIA的宏蛋白质组学方法的还不被更多人熟悉,这正是我们这篇最新发表的文章期望传达的主要信息,进而能够提供支撑信息,让这个技术带来的优势被更多人了解。
DIA改变了宏蛋白质组学数据的生成方式
传统的基于DDA的蛋白质组学分析是这样的:MS2采集由MS1扫描中的母离子峰触发(换句话说:“依赖于”MS1扫描)。蛋白质鉴定是通过在MS2谱图中搜索肽片段来实现的,因此,如果没有选择肽前体进行MS2片段化,肽前体的信息将会丢失。更糟糕的是,随机前体选择可能导致“缺失值”问题,与传统的单一生物体蛋白质组学相比,这在宏蛋白质组学中更为显著,因为样品异质性、复杂性和动态范围显著增强。
相比之下,DIA在一个采集周期内执行一系列MS2扫描,其中定义的隔离窗口内的母离子可无遗漏的采集并多次碎裂。此功能允许系统无偏地记录样品中理论上存在的所有肽的碎裂信息。甚至可以略夸张地说,样品多肽信息已被数字化记录以进行长期存储。未来使用更先进的信息学解决方案进行再分析可能有助于获得目前无法检测到的生物学发现。易算已经重分析了一系列数据证明了这一点:directDIA+为您点亮质谱数据中的黑暗角落
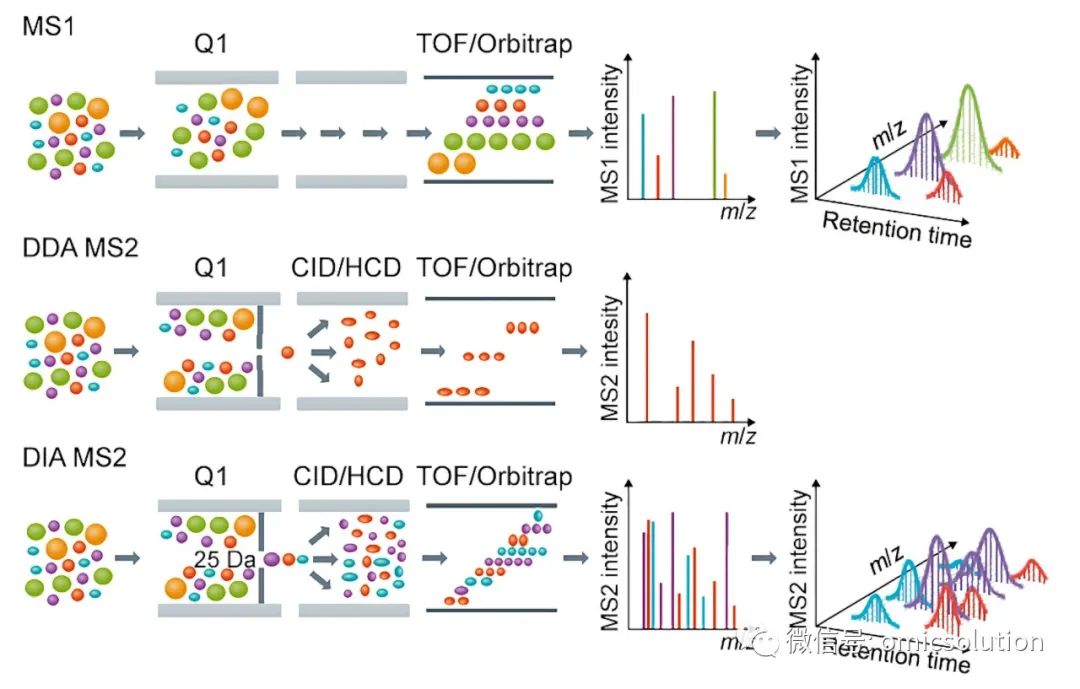
图1.DDA 和 DIA 方法的示意图。
改编自 CC BY 4.0 许可下的参考文献 1
DIA加深了宏蛋白质组的覆盖率,提高了定量准确性
那么有人就要说了:除 DIA 外,不要忘了常用的定量策略包括基于 DDA 的无标记定量 (LFQ) 和串联质量标签 (TMT) 等重异位标记定量。这些技术也很好啊。在典型的LFQ-DDA分析中,蛋白质定量基于母离子强度。在TMT分析中,蛋白质定量基于等重异位标记MS2碎裂后产生的报告离子。
数字说话。
本文,我们对掺入6种已知细菌物种的人粪便代谢蛋白质组样品的DIA和其他两种定量策略进行了基准测试。相较于LFQ-DDA,DIA具有卓越的蛋白质组覆盖率和数据完整性。与LFQ-DDA相比,DIA产生的蛋白质和肽多出约80%和55%,这些蛋白质被有效鉴定和定量(缺失值不超过1/3)。
我们进一步从定量的蛋白质中,选择属于这6个物种的蛋白质来评估方法的定量准确性。我们证明了DIA在要求更高的定量准确性方面优于TMT。在极度复杂样品中DIA的定量结果准确性更是优于简单混合物样品以及模拟12个物种的微生物群落。(我们非常感谢审稿人关于模拟真实宏蛋白质组样品复杂性的建议)。
为什么有如此大的区别?
该区别来自复杂宏蛋白质组样品中共分离和共碎裂肽的报告离子强度叠加,TMT遭受比率压缩,测量的倍数变化严重偏离理论值,间接证明了在许多benchmark测试中得到的结论并不能直接用于实际的复杂生物学样品中。而DIA可以根据肽特异性MS2色谱图XIC信息进行蛋白质定量,该信息可以有效减轻其他肽的干扰,从而提高定量准确性。
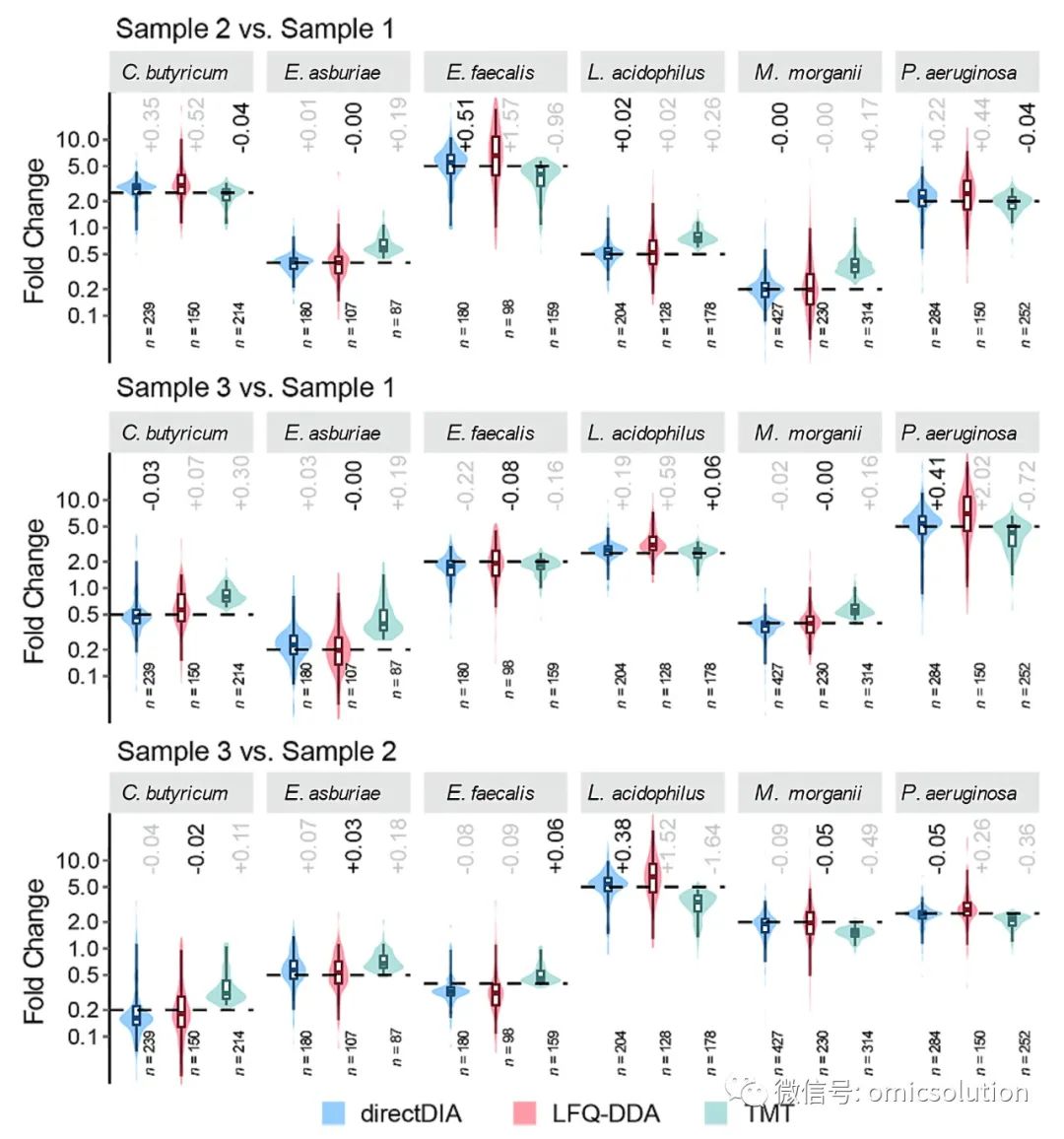
图2.DIA、LFQ-DDA和TMT对加标宏蛋白质组样品的定量准确性比较。
DIA定量不再必须使用谱图库
很多人会有一个误解:DIA的一个主要缺点是其数据分析需要一个谱图库,该谱库通常由DDA分馏样品构建而成。
真的是这样吗?最近,Pietilä等人引入了用于宏蛋白质组学分析的无谱图库DIA【5】。通过结合深度学习改进信息解决方案【6】,DIA 分析不再必须使用实验谱图库。
在本文中,我们将Spectronaut中的无谱图库工作流程directDIA与基于实验和预测光谱库的替代DIA数据分析方法进行了比较。结果表明,directDIA在微生物群落的复杂蛋白质组样本上优于基于谱图库的方法。
directDIA数据分析的唯一所需先验知识是蛋白质序列数据库。我们应用directDIA在两个真实的临床队列中表征人类肠道微生物群。结果表明,directDIA与主流的宏蛋白质组序列数据库选择配合良好【7】,即样品的实验宏基因组和由公开可用的序列组装的“pseudo-宏基因组”。
单物种谱图库显著提升混合菌定量深度
看到这里也许就有人会问了:“那谱图库是不是就没啥用了呢?”
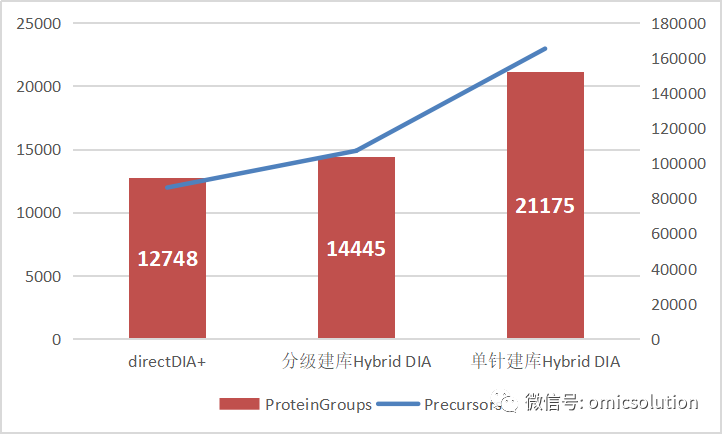
在新版本Spectronaut17支持hybrid(也就是directDIA+DDA分级谱图库联合分析功能)后,我们进一步测试了该策略的效果。从图3可以看到,混合建库大约提升了10%的定量结果。从性价比来说属于锦上添花的效果而非原先的雪中送炭了。
|
directDIA+对比hybrid-DIA定量
图3.directDIA+对比Hybrid定量结果 |

不过,本实验相对于宏蛋白质组实际实验有非常大的区别:
可以获得单一物种细菌群落;
单一物种细菌蛋白质组相对简单。
那么我们为啥非得把简单的细菌混合成复杂样品去分级建库呢?
于是,我们进一步在最新的timsTOF Pro2上将每种细菌分别进行了单针DIAPASEF检测并以此建库。(实际样品由于无法获取单一物种群落,并不可行。)
|
单物种谱图库
图4.基于单针DIA的谱图库构建结果 |
|
|
|
|
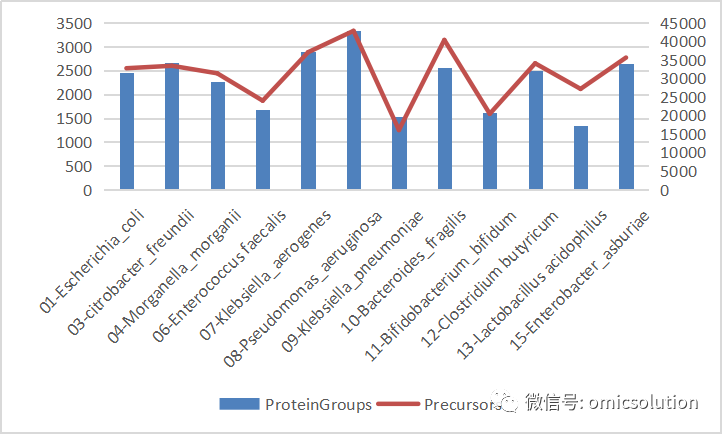
从图5看似乎单针谱图库和分级比差不少。那实际情况如何呢?
图6展示了和文献相同的12个细菌混合物单针DIA定量的结果数量,分别采用了directDIA+,DDA混合分级和DIA单物种单针建库。单针单物种建库的谱图库覆盖率高达90%(Protein Groups),74%(Precursors),从而实现单针1小时DIAPASEF定量超过21000种Protein Group(species specific)的超高覆盖度。同时,由于precursor水平的极高覆盖率也就是蛋白质的序列覆盖率的极大提高,宏蛋白质组中物种区分难的问题在一定程度上可以得到改善(species specific 肽的检测成功率提高)。
虽然该策略无法直接用于宏蛋白质组实际样品定量,但对于有目标细菌物种定量或者类似需求的动物细胞、组织定量,有着极佳的参考价值。
总结
DIA在肠道微生物群的深度表征中具有广阔的前景。随着仪器进一步创新,Spectronaut等软件的进一步迭代,DIA宏蛋白质组学必将更加通用,而且大放异彩。尤其是宏蛋白质组中的准确蛋白质鉴定和定量这类卡脖子的问题一旦取得了飞跃性的进步,新的可能性才会出现。
好了,以上就是今天的内容。假如你正在或者将要开展DIA宏蛋白质组学方面的课题,想找到更多更有价值的信息,请你和我们联系。假如您的朋友目前有开展宏蛋白组课题的想法,想深入了解相关数据质量,也请你帮我们把这篇文章转发给他。
- 参考文献简介
1. Ludwig, C. et al. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14, e8126 (2018).2. Aakko, J. et al. Data-independent acquisition mass spectrometry in metaproteomics of gut microbiota—implementation and computational analysis. J. Proteome Res. 19, 432-436 (2020).3. Long, S. et al. Metaproteomics characterizes human gut microbiome function in colorectal cancer. npj Biofilms Microbiomes 6, 14 (2020).4. Muntel, J. et al. Comparison of protein quantification in a complex background by DIA and TMT workflows with fixed instrument time. J. Proteome Res. 18, 1340-1351 (2019).5. Pietilä, S., Suomi, T. & Elo, L. L. Introducing untargeted data-independent acquisition for metaproteomics of complex microbial samples. ISME Commun. 2, 51 (2022).6. Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41-44 (2020).7. Tanca, A. et al. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome 4, 51 (2016).